Genome Editing by the CRISPR/Cas9 System
Introduction
What is CRISP?
ClusteredRegularly Interspaced Short Palindromic Repeats (CRISPR) along with its well-defined CRISPR associated protein (Cas) are recognised to be a powerful bacterial defence mechanism against invading bacterial phages. Since this natural process was first discovered, researchers have been quick to recognise its potential as a research tool. The CRISPR/Cas9 system is now extensively used as a more effective technique for genome editing than previous versions of the technology such as zinc finger nucleases (ZFN), Transcription activator-like effector nucleases (TALEN) and small interfering RNA (siRNA), which required researchers to design and produce specific nuclease pairs for each experiment. This introduction defines the fundamental components of the CRISPR/Cas system, the guide RNA (gRNA) and the Cas endonuclease, and describes the variations which can be used for different genome editing objectives.
How does CRISPR work?
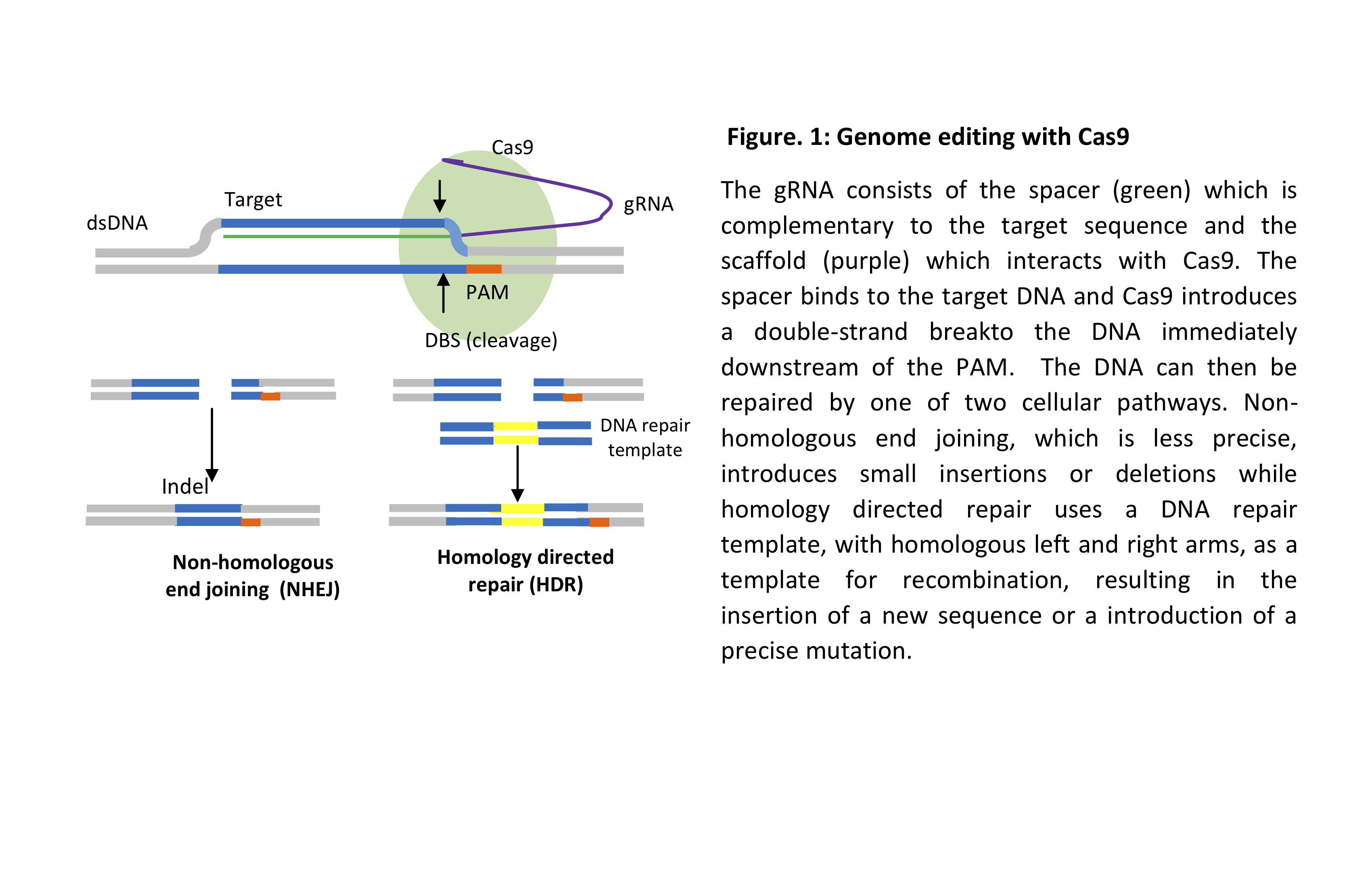
In bacteria, the gRNA, which carries a segment of a previously invaded phage, binds to the endonuclease protein Cas9 via a scaffold sequence. It then binds to the complementary sequence in the genome of a newly invaded phage, upstream of the protospacer adjacent motif (PAM) sequence NGG. Cas9 then makes a double-strand break (DSB) and the cleaved DNA is then degraded, thus conferring resistance to the invading phage. Genome editing follows the same principle by designing a gRNA complementary to the gene of interest and selecting a Cas protein based on the objective of the experiment, for example, gene inactivation or sequence editing (Figure 1) as well as activation or repression of gene expression as well as other applications.
Uses for CRISPR genome editing experiments
Selection of the most suitable method or workflow for a genome editing experiment is dependent on the purpose of the research, from permanently silencing, activating, or controlling the expression of a gene of interest to causing a frame shift mutation, reversing an existing mutation, visualising a specific gene of interest or even screening an entire library for a set of genes. The following experiments can be done with CRISPR, each with a specific method and products.
Method and specific products.
Knock-out
In knock-out experiments, CRISPR is likely causing the complete loss of function of a certain gene as it depends on the cellular mechanism of non-homologous end joining (NHEJ) which repairs double-strand breaks (DSB) introduced into the genome. This non-precise method of DNA repair results in small insertions or deletions (Indels) which change the original sequence of the gene, resulting in loss of function, see Figure 1.
Knock-In
In an editing experiment a DNA repair template with the desired edit flanked by homologous sequence termed left and right homology arms, is introduced into the cell along with the gRNA and Cas endonuclease and relies on the cellular mechanism of homology directed repair (HDR). A double-strand break is introduced upstream of the PAM, then the DNA repair template is used to repair the DSB thus introducing the sequence of interest. The DNA repair template can be introduced as a single-stranded oligonucleotide, a double-stranded oligonucleotide or plasmid. The edited sequence can be in the form of a point mutation, the replacement of a weak promoter with another to enhance the expression of a gene or the reversal of an existing mutation are just a few examples. Recombinant versions of the Cas9 protein can introduce single-strand breaks, see Figure 1b.
CRISPR interference (CRISPRi)
While knock-out experiments can be achieved by altering the coding sequence of a gene to disable it, gene expression can also be controlled without altering the target sequence. The introduction of certain point mutations to the Cas9 gene sequence has led to the creation of a mutated protein know as dead Cas9 or dCas9, which can bind to its target without inducing DNA cleavage. This binding, however, interferes the expression of the target gene by preventing transcription factors from accessing the gene, therefore silencing its expression. A transcriptional repressor protein can be fused to dCas9, allowing a reversible and fine-tuned reduction in gene expression rather than complete silencing. Using dCas9 for interference has advantages over the use of siRNA, for example, it decreases off-target effects and increases the on-target effect.
CRISPR activation
In addition to gene repression, a transcriptional activator can also be fused to dCas9 for the purpose of up-regulating gene expression
Gene Visualisation
CRISPR can also be used to visualise genomic regions by attaching fluorescent proteins, for example, green fluorescent protein to dCas9 proteins and using the CRISPR system to tag desired region of the genome.
CRISPR screens
With recent developments in CRISPR, genomic screening libraries are now available in order to knock out thousands of genes in a single screen with high efficiency. This procedure has also been used before for studying the various functions and phenotypes of genes by the more commonly used siRNA method.
Different Formats for Introducing CRISPR into the Cell
As mentioned before the two fundamental components of CRISPR/Cas9 system are the guide RNA and the CRISPR associated endonuclease protein. These two components can be introduced into the cell in different formats.
DNA Format
In this format, the gRNA and Cas9 protein are cloned as a synthetic DNA fragment into a plasmid vector via the standard cloning workflow. The first step involves designing the DNA template to encode the gRNA and Cas9 protein. Once the DNA template sequence has been designed, an oligo can be ordered and cloned into a plasmid. After selection and confirmation of the presence of the desired fragments, the recombinant plasmid is then transfected into the cells of the experiment. The cells express the gRNA and Cas9 which then target the gene of interest. The complexity of accurately designing the necessary sequence, the time-consuming workflow of the cloning steps and the potential high error rate make this format the most difficult and the least efficient for CRISPR delivery. Even with commercially available recombinant plasmids, this format still has limitations. Plasmids are difficult to introduce into cells due to their large size compared to other delivery forms. There is also a possibility that the plasmid will integrate into the host genome in places that cause cytotoxicity and the continuous presence of the Cas9 protein potentially increases the rate of off-target effects.
In this format, the CRISPR components are delivered as guide RNA and mRNA encoding the Cas9 protein. The process involvesin vitrotranscription of the DNA template containing the sequence of gene of interest prior to introduction into the cell. An upstream promoter site is typically incorporated into the design for RNA polymerase to use for in vitrotranscription. Commercially formulated kits are available for this purpose, yet it is still costly with the potential to introduce errors. After purification, the gRNA can be co-transfected into the cell alongside the Cas9 mRNA.
Synthetic guide RNA
This format is a convenient substitute to the in vitrotranscription-derived guide RNA and involves the synthetic polymerisation of high-quality RNA (up to 120-mer) sequences. These can either be in the form of separate CRISPR RNA (crRNA or spacer) and trans-activating crRNA (tracrRNA or scaffold) fragments that must be annealed together, much like the natural bacterial system, or as seamless single guide RNA (sgRNA). Of these two forms, sgRNA generally produces a higher editing efficiency by avoiding the inefficient annealing of the two-piece system, and the tendency of tracrRNA fragments to form tetramers that interfere with the Cas9 protein.
Ribonucleoprotein (RNP)
Dimerising sgRNA with Cas9 as a ribonucleoprotein complex is the most effective strategy, as researchers have demonstrated higher editing efficiencies and fewer potential off-target effects using RNPs compared to other delivery methods. Moreover, as the RNP exists transiently within the cell and is not inserted into the genome, there is a low risk of cytotoxicity. It has become clear that the RNP format is the most effective option for performing CRISPR experiments, especially in embryos.
gRNA
The gRNA is a key element in CRISPR system and should be designed carefully to ensure maximum on-target effect i.e. targeting only the genomic region of interest and minimise off-target effects which may occur if there is partial homology with other sequences in the genome. The selected application must also be taken into consideration when designing the gRNA.
For Knock-out experiments:
- Choose guide sequences which target exons that are present in all transcript variants.
- Choose sequences at the 5’ end of the gene or that encode essential protein domains as these are more likely to interfere with protein function.
For knock in, CRISPRi and CRISPR activation (CRISPRa) gRNAs have additional requirements. For knock-in experiments, the template DNA should be carefully checked for PAM sequences to ensure that they are not treated as a target. For CRISPRa/ CRISPRi applications, the gRNA sequence should be complementary to the promoter regulating the target gene, rather to the sequence of the gene itself.
Design tools
The team at Willowfort will design the CRISPR gRNA for you as part of the service, accompanying this kit. If you require further information, there are plenty of online CRISPR design tools now available.
Cas proteins
Cas endonuclease is the cutting proteins in the CRISPR system. There are different types and choosing one is dependent upon the application in question. Not only are there the naturally occurring Cas proteins, but also one with mutated domains has been created to suit more applications.
- pyogenes Cas9 (SpCas9): the bacterial strain Streptococcus pyogenesintroduces the most commonly known and used Cas9 nuclease. This nuclease recognises the PAM motif 5’-NGG-3’ and makes a double-strand break leaving blunt ends at the target sequence. It was the first nuclease engineered for CRISPR experiments and is used for a variety of knockout applications.
- aureus Cas9 (SaCas9): the bacterial strain Staphylococcus aureusintroduces an ortholog of the Cas9 family. SaCas9 recognises the PAM sequence 5’-NNGRRT-3’. This protein is much smaller than SpCas9, allowing it to be packaged into an AAV vector for cellular delivery, an approach that is difficult with the larger SpCas9.
- Cpf1: functionally differs from SpCas9 in three key aspects:
- It recognises the PAM motif 5’-TTN-3’. It can therefore be a better choice for
targeting DNA regions with high AT-content than Cas9.
- It creates sticky ends rather than the blunt ends of SpCas9. Thus Cpf1
is preferred for experiments relying on the HDR repair outcome.
- It is a smaller protein than SpCas9 and does not require a tracrRNA. Thus, the gRNA required by Cpf1 is shorter in length and cheaper to generate than the gRNA required by SpCas9.
- Cas9 Nickase:a modified version of the Cas9 protein which nicks a single DNA strand. Additionally, this nuclease makes a staggered cut that leaves long overhangs instead of blunt ends at the cleavage site. This enables more control when inserting a DNA segment for HDR repair. For this reason, Cas9 nickase is often used for knock-in experiments.
- dead Cas9 (dCas9): is a mutated Cas9 protein which binds to the target sequence but does not cleave it. Instead, it blocks the cellular transcription mechanism from expressing the target gene, thus silencing gene expression without introducing breaks in the DNA. It is similar to the action of siRNA but with much more on-target effect and the less off-target effects. It is likely to be used in CRISPRi and CRISPR experiments.
Transfection of CRISPR components into the cell
One of the key success factors in any CRISPR experiment is the introduction of the CRISPR components into the cell line. Choosing the right mode of transfection is dependent on many factors including the cell type itself and the format of the CRISPR system.
Four well-defined methods for transfection are as follows:
Lipofection: is a lipid-based transfection technology which associates nucleic acids with cationic lipid formulation. The resulting molecular complexes, known as lipoplexes, are then taken up by the cells. When applying lipid-based transfection reagents, there are three general measures of an effective transfection reagent:
- High-efficiency delivery
- Low cellular toxicity
- Broad dynamic range
Lipofectamine is a common transfection reagent, which is used to increase the transfection efficiency of RNA, including mRNA and siRNA or plasmid DNA into vitrocell cultures.
Electroporation & Nucleofection
Electroporation enables delivery of the CRISPR components into cell types that are difficult to transform using lipid-based delivery systems. Delivery of a controlled, short electric pulse to the cells forms pores in the cell membrane, allowing entry of foreign material. Nucleofection is a variant of electroporation, in which the electric pulse is optimised to induce pore formation in the nuclear membrane as well, thus delivering the CRISPR components directly into the nucleus.
Microinjection
This is commonly used to inject the Cas9 and gRNA ribonucleoprotein complex into embryos, although it can also be used in cells. Zebrafish, mouse and most recently human embryos have been manipulated using this technique.
Lentiviral transduction
Lentiviral vectors derived from the human immunodeficiency virus (HIV-1) have become major tools for gene delivery in mammalian cells. The advantageous feature of lentivirus vectors is the ability to mediate potent transduction and stable expression into dividing and non-dividing cells both in Vitro and in vivo, it can also be used with hard-to-transfect cells.
Genome Editing CRISPR/Cas9 (Knock out) complete kit
Overview
The innovative Genome Editing CRISPR/Cas9 (knockout) Complete Kit is formulated for use in a typical knockout experiment with the highest efficiency (on-target effect) and the least off-target effects. It contains all the necessary components to complete the experiment in a convenient way. The highly purified SpCas9 is introduced in its protein form and both crRNA-tracrRNA are introduced in their sgRNA form. The protocol utilises the RNP formation step before transfecting the cells to ensure maximum product efficiency.
Features
Efficient–Simple and quick method for transcribing high yields of sgRNA in the one tube.
Reliable– Willowfort kits undergo extensive tests to ensure consistent quality in all of our products.
Robust– High performance and quality of synthetic CRISPR gRNA greatly increased relative to in-vitro transcribed gRNA & plasmid mechanism.
Research use only.
Experimental Workflow
The following is a general protocol to deliver SpCas9 nuclease protein NLS and RNP complex into cultured mammalian cells. Exact reagent amounts and parameters for co-transfection should be empirically determined through careful optimisation in your cells of interest prior to experimentation. Reagent volumes can be calculated to include replicate samples as necessary. Four different types of samples are recommended for a gene editing experiment as follows:
- Positive control samples: SpCas9 protein with tracrRNA and an undesired targeting guide RNA
- Negative control sample: SpCas9 protein with tracrRNA and non-targeting control guide RNA.
- Gene editing sample: expression of Cas9 protein and gRNA for targeted double-strand cleavage in the gene of interest).
- Untransfected: no treatment control sample (confirmation of cell viability)
Why use both negative & positive controls?
|
Positive control |
Description | Function |
| Pre-designed crRNA which targets an undesired sequence, usually a housekeeping gene.
Provided with primers to proceed mismatch cleavage assay.
|
Shows that Cas9 editing is functional in the cells.
Can also serve as a negative phenotypic control for screens where gene editing without a resulting phenotype is desired.
|
|
| Negative control | Pre-designed crRNA not known to target any human, mouse or rat genes. | Used to determine the difference between the cellular responses to the presence of any gRNA and the effects of a gene-specific gRNA |
Transfection
All steps of this protocol should be performed in a laminar flow cell culture hood using sterile technique. Wear a lab coat, gloves and safety goggles. Dispose of waste according to local and national regulations.
- Setting up the 96 well plate
- Dilute the cells in antibiotic-free complete medium to the appropriate stock density. For example, 10,000 cells in 0.1 mL of medium for plating at 10,000 cells/well in a 96-well plate. For the whole plate, make up a solution of 1×106cells in 10 mL.
- Plate 0.1 mL of cell suspension into each well of a 96 well plate and incubate the cells at 37 °C with 5% CO2 overnight.
- Reagent preparation
2.1 Prepare a stock solution of SpCas9 protein in the vial provided by resuspending in 30 µl of 1X Resuspension Buffer (10mM Tris,300mM NaCl,0.1mM EDTA,1mM DTT, pH7.4, 50% glycerol) to achieve a concentration of approximately 17mg/ml (10 pmol/µL).
2.2 Allow the solution to incubate for at least 10 minutes at room temperature (20-23°C) to completely dissolve in solution. Vortex and centrifuge immediately before use.
2.3 If a lower concentration of Cas9 protein is required, dilute the Cas9 protein with the supplied Dilution Buffer (20 mM Na-HEPES pH 7.5, 200 mM NaCl) immediately before use. Store the diluted protein on ice for up to 6 hours.
- RNP complex formation
3.1 Assemble RNP complexes on the ice, immediately before transfection as follows:
In a sterile nuclease free tube prepare the RNP complex in an equal molarity ratio i.e. 1:1 sgRNA: Cas9 protein in a volume of 50ml per well.
E.g. 1.5 µL of 20 µM sgRNA, 1.5 µL of 20 µM SpCas9 protein and 47µl of
Opti-MEM (serum free medium) per well multiplied by the number of wells being used. Some optimisation may be required.
3.2 Incubate this mixture at room temperature for 5 minutes to assemble the RNP complexes.
3.3 To further optimise, incubation on ice for up to 30 minutes.
The RNP complex can be stored for up to 4 weeks at –4°C or –20°C.
- Lipofection optimisation protocol
The following factors have the greatest impact on transfection efficiency and need to be optimised for the particular cell type used:
4.1 Poor cell health and high passage number can negatively affect lipofection. Use the lowest passage number possible for your cells, especially if you observe significant cell death during transfection.
4.2 Lipofection requires dividing cells. Cell density at the time of transfection should be sub-confluent, typically 60–75%. Low confluency can lead to higher cell death and high confluency may affect transfection efficiency.
4.3 The amount and the relative ratio of lipid to RNP complex: for optimisation of the 96-well plate, vary the ranges of the following components:
| Component | Optimisation range |
| CRISPR-Cas9 RNP complex | 3, 10, and 30 nM |
| Lipofection Kit | 0.2–2.0 μL |
- Transfection mixture preparation
5.1 Add 50 µL of the provided Lipofection to each RNP complex tube to give a final volume of 100 µL. The volume of Lipofection should be optimised for each cell type.
5.2 Pipet up-and-down gently to mix.
5.3 Incubate at room temperature for 15-30 minutes to allow transfection complex formation.
- Cell transfection
6.1 Distribute transfection complexes to the cells in complete growth medium by removing medium from the wells of the 96-well plate containing the cells and replacing it with 100 μL of the appropriate transfection medium.
6.2 Incubate the cells at 37°C with 5% CO2for 14-18 hours then replace the transfection mix on the cells with regular growth medium without antibiotics.
6.3 Incubate the cells at 37°C with 5% CO2for 72 hours after the initial transfection, before proceeding with gene editing analysis.
Determining Genome Targeting Efficiency using T7 Endonuclease I
T7 Endonuclease I recognises and cleaves mismatched DNA. This protocol describes how to determine genome targeting efficiency by digesting annealed PCR products with T7 Endonuclease I. In the first step PCR products are produced from the genomic DNA of cells which were targeted using CRISPR/Cas9. In the second step, the PCR products are annealed and digested with T7 Endonuclease I. Fragments are analysed on a gel to determine the efficiency of genome targeting.
- PCR
1.1 Set up a 50 µl PCR reaction using approximately 100 ng of genomic DNA as a template.
For each amplicon set up reactions in triplicate using the following templates:
- gDNA from Cas9 transfected cells
- gDNA from negative control cells (i.e. non-specific DNA transfected cells)
- water (no template control)
- Components of PCR using High-Fidelity DNA Polymerase
| Component | 50 µl Reaction | Final Concentration |
| Cosmo Hot Start High-Fidelity 2X Master Mix | 25 µl | 1X |
| 10 µM Forward Primer | 2.5 µl | 0.5 µM |
| 10 µM Reverse Primer | 2.5 µl | 0.5 µM |
| Template DNA | variable | 100 ng total |
| Nuclease-free water | To 50 µl |
1.3 Gently mix the reaction. Briefly centrifuge to collect all the liquid at the bottom. Transfer the PCR tubes to a PCR machine and begin thermocycling.
1.4 Cycling Conditions
| Step | Temperature | Time |
| Initial Denaturation | 95°C | 120 seconds |
| 35 cycles | 95°C | 10 seconds |
| *50-72°C | 20 seconds | |
| 72°C | 20 seconds | |
| Final Extension | 72°C | 2 minutes |
| Hold | 4-10°C |
*Depends on the melting temperature (Tm) of the primers
- Purification of the PCR product
Note: all centrifugation steps should be performed between 10,000-16,000 x g. Willowfort recommends the use of its Nova PCR Clean-up Kit for this procedure.
2.1 In a 1.5 ml microcentrifuge tube, add DNA Binding Buffer to each of DNA samples in the following ratio 5:1 DNA Binding Buffer: Sample, e.g. 500 mL: 100 mL. Mix briefly by vortexing.
2.2 Transfer the mixture to a provided Spin Column in a Collection Tube.
2.3 Centrifuge for 30 seconds. Discard the flow-through.
2.4 Add 200 µl DNA Wash Buffer to the column. Centrifuge at for 30 seconds, then discard the flow-through. Repeat the wash step.
2.5 Add a minimum of 25 µl DNA Elution Buffer directly to the column matrix and incubate at room temperature for one minute.
2.6 Transfer the column to a 1.5 ml microcentrifuge tube and centrifuge for 30 seconds to elute the DNA.
- T7 Endonuclease I digestion
3.1 Assemble the annealing reactions as follows:
| Component | Amount |
| DNA | 200 ng |
| 10X Buffer | 2 µl |
| Nuclease-free Water | To 19 µl |
3.2 Anneal the PCR products in a thermocycler using the following conditions:
Annealing Conditions:
| Step | Temperature | Ramp Rate | Time |
| Initial Denaturation | 95°C | 5 minutes | |
| Annealing | 95-85°C | -2°C/second | |
| 85-25°C | -0.1°C/second | ||
| Hold | 4°C | Hold |
3.3 Add T7 Endonuclease I to the annealed PCR products as follows:
| Component | Amount |
| Annealed PCR product | 19 µl |
| T7 Endonuclease I Enzyme | 1 µl |
| Incubation Time | 20 minutes |
| Incubation Temperature | 37°C |
3.4 Stop the reaction by adding 1.5 µl of 0.25 M EDTA.
3.5 Analyse the fragmented PCR products on an agarose gel and determine the percent of nuclease-specific cleavage products (fraction cleaved).
Calculate the estimated percentage of gene modification using the following formula:
% gene modification = 100 x (1 – (1- fraction cleaved)1/2)
Troubleshooting
When it comes to CRISPR experiments, the optimisation of reagents and experiment layout design are as important as the experiment itself. At least 15% of CRISPR experiments fail for obvious reason but the creation of variability between the experiments in one of the major factors, for example, different Lipofection kit volumes, however, the inclusion of positive and negative different controls may help in understanding the reason for experiment failure.
Here are the most common reasons for CRISPR experiment failure, although other reasons less common reason may also contribute to this.
Observed problem |
Possible cause |
|
Cell death after transfection
|
Widely observed cell death following transfection is an indication that the lipofection optimisation protocol needs to be re-optimised to match the cells of the experiment. These are some of the variables
· Amount of transfection reagent · The lot/batch of transfection reagent · Duration of transfection · Cell density at transfection
If the experiment has been done without varying volumes of lipofection, it is strongly recommended to repeat it with at least 5 different volumes of lipofection to determine which one is the most optimal for the used cells. Often decreasing the amount of transfection reagent and/or the total duration of transfection will help minimise the toxic effect on the cells. |
|
Mismatch analysis shows low percentage of editing |
· Mismatch DNA endonuclease digestion can lead to non-specific cuts that degrade the PCR product and reduce the intensity of the desired bands, especially at longer incubation times.
· If the CRISPR-Cas9 gene editing generates large inserts or deletions, primer binding sites can be impacted and the mutation thus will not be detected by the DNA mismatch assay.
|
| No knockout observed | This problem in particular includes many factors:
· Unsuccessful delivery of CRISPR/Cas9 components. · Wrong choice of the specific Cas protein · Very low concentrations of Cas9 protein are used · Inactivated Cas9 (Cas9 is still bound to the target DNA even after cleavage) · Poor guide RNA design
|
Product Listing
|
Product |
Package Size |
Order Information |
|
CRISPR-Cas9 crRNA |
2 nmol | WC90102002 |
|
10 nmol |
WC90102010 |
|
| CRISPR-Cas9 tracrRNA |
2 nmol |
WC90103002 |
|
10 nmol |
WC90103010 |
|
| S. Pyogenes Cas9 Nuclease |
100 µg |
WC90104010 |
| S. Pyogenes Cas9 Nickase |
100 µg |
WC90105010 |
| S. Pyogenes Cas9 Protein | 100 µg |
WC90106010 |
|
Learn more about our Custom CRISPR Cas9 Knockout Kit |
WC9010xxxx010 |
|
|
Learn more about our custom CRISPR Cas9 Vector |
WC90V40xxxx010 |
|
| Target sequence cloning into CRISPR-Cas9 Vector |
WC90QV74xxxxx01 |
|
|
CRISPR-Cas9 Control Kits |
20 rxn |
WC90109010 |
|
Cas9 Nuclease Reaction Buffer 10X |
5 x 1.5ml |
WC90111005 |
Select from our Predesign gRNA
FAQs
- What is the optimal length of the homology arms, when designing a DNA repair template for HDR? What is the maximum size of the donor insert?
In general, the length of the left and right homology arms is dependant upon the size of the donor insert, the longer the insert, the longer the homology arms need to be. A commonly used length is approximately 500bp for each homology arm in mammalian cells and approximately 1-2kb for the insert range. If a single-stranded oligo DNA (ssODN) is used, the homology arms can be as little as 40bp and although the length of ssODNs were once limited to approximately 200bp, ssODNs of up to 1kb have been reported. Longer inserts and homology arms can decrease the efficiency of recombination.
References:
- Thomas KR, Folger KR, et al. (1986) High frequency targeting of genes to specific sites in the mammalian genome. Cell. 44(3):419–428.
- Dickinson DJ, Ward JD, et al. (2013) Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature methods. 10(10):1028–1034.
- Yoshimi K, Kunihiro Y, Kaneko T, Nagahora H, Voigt B, Mashimo T, (2016).
ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat Commun. 20(7):10431
- Will the PAM sequence be included in the design of the sgRNA?
The PAM sequence will not be included in the sgRNA because this will interfere with normal Cas9 function. The PAM sequence should be located on the non-target strand, immediately downstream of the DNA target, see Figure 1.
Reference:
Anders C, Niewoehner O, et al. (2014) Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 513(7519):569–573.
- What is the average frequency of PAM sequences in the mammalian genome?
The NGG motif makes up approximately 5.21% of the human reference genome, therefore there is a ‘GG’ dinucleotide every 42 nucleotides. If a PAM sequence falls within the proposed DNA repair template, one of the nucleotides can be changed to a nucleotide that will give a synonymous change, i.e. one that does not result in a change in amino acid.
Reference:
Scherer S Cold Spring Harbor Laboratory. Press., A short guide to the human genome2008, Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press. xiv, 173
- Can the CRISPR/Cas system be used to simultaneously target multiple loci in the genome?
Several approaches have been used to multiplex CRISPR/Cas9 targeting. In one paper plasmids containing 3 sgRNAs and Cas9 were co-transfected into ES cell lines, resulting in all 6 alleles being altered. A further approach was to target 5 genes with short PCR products of the sgRNAs were co-transfected with the plasmid containing Cas9. Ten percent of clones contained all 6 mutated alleles (2 genes were on the Y chromosome). The percentage of clones, in this experiment, containing the 3 genes originally delivered as plasmids was higher which is probably due to the PCR products being smaller than plasmids [1]. Another application used Cas9 and CRISPRainbow to visualise 6 genomic loci. RNA hairpins that recruit fluorescent proteins were fused to sgRNA targeting telomeres and repeat sequences, with combinations of primary coloured fluorescent proteins producing up to seven different colours including white. The gRNA hybridised to their targets to fluorescently tag the target repeat sequences [2]. A polycistronic plasmid containing multiple target sequences has been used in Saccharomyces cerevisiae[3]. CRISPR libraries, containing gRNAs which are cloned into lentiviral vectors, are now available commercially. These libraries can potentially target a class of genes or even every gene in the genome [4].
References:
- Wang H, Yang H, et al. (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 153(4):910–918
- Ma H, Tu LC, Naseri A, Huisman M, Zhang S, Grunwald D, Pederson T, (2016) Multiplexed labelling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat Biotechnol. 34(5):528-30.
- Bao Z, Xiao H, et al. (2014) Homology-Integrated CRISPR-Cas (HI-CRISPR) System for One-Step Multigene Disruption in Saccharomyces cerevisiae. ACS synthetic biology.
- McDade JR, Waxmonsky NC, Swanson LE, Fan M, (2016) Practical Considerations for Using Pooled Lentiviral CRISPR Libraries. Curr Protoc Mol Biol. 1(115):31.5.1-31.5.13
5. How can off-target effects be minimised?
Ideally, the gRNA will only match the intended target, termed the on-target effect, however it is likely that there will be some homology with other sites in the genome (off-target). If the gRNA has mismatches in the sequence close or adjacent to the PAM at these off-target sites, the cleavage efficiency will be greatly reduced. The software is available which aims to minimise off-target effects such as those previously mentioned.
- Following a genome editing experiment, how can I check the on-and off-target effects?
T7 endonuclease I is included in this kit and a protocol is included on page 18 of this CRISPR in order to assess the genome targeting efficiency. The endonuclease targets mismatch heteroduplexes in PCR products and cleaves them. The products are run on a gel.
As previously mentioned, Sanger sequencing or next generation whole-genome sequencing can also be used. The reference below also gives other methods for determining off-target effects.
Reference:
Hendel A, Fine EJ, et al. (2015) Quantifying on- and off-target genome editing. Trends in biotechnology. 33(2):132–140.